新聞中心 / News Center
貝達藥業多項自主研發成果亮相AACR年會
日期: 2022-04-15

4月8日-13日,第113屆美國癌癥研究協會(AACR)年會在美國新奧爾良舉行。貝達藥業鹽酸埃克替尼術后輔助治療、恩沙替尼MET 14跳躍突變探索、BPI-361175、BPI-371153、BPI-421286、BPI-442096等多項自主研發成果在會議上亮相。
01
評估埃克替尼輔助治療IIA-IIIA期EGFR突變非小細胞肺腺癌療效的多中心、開放標簽、單臂、II期研究(ICAPE)
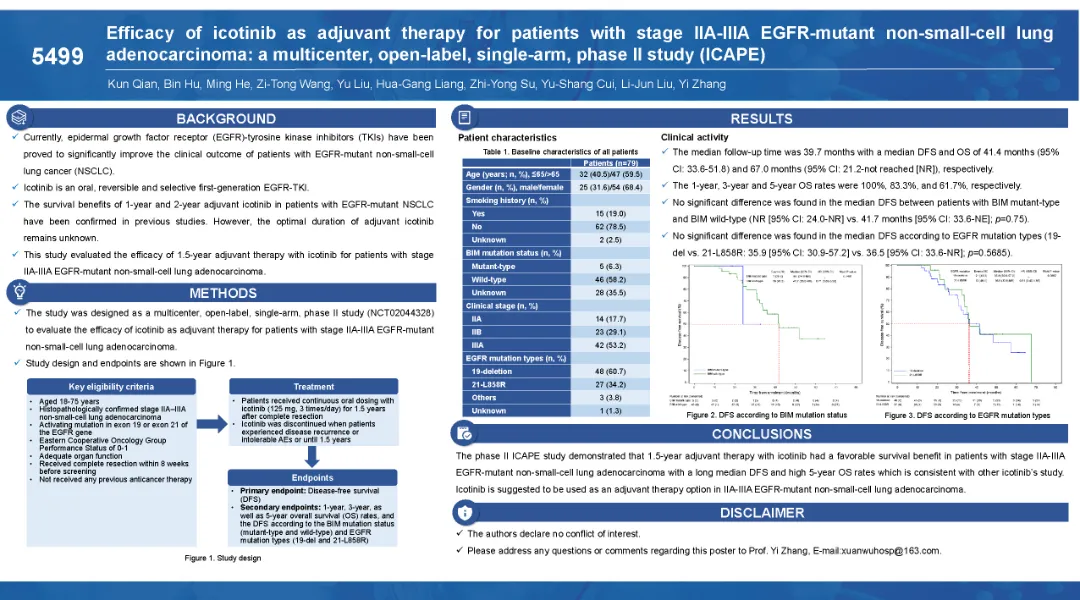
多項研究證實了埃克替尼在晚期肺癌患者中的生存獲益。本研究(ICAPE)旨在評估埃克替尼作為輔助治療II-IIIA期表皮生長因子受體(EGFR)突變型非小細胞肺腺癌患者的療效。
本研究納入II-IIIA期EGFR突變的非小細胞肺腺癌患者。符合條件的患者在完全手術切除后接受口服埃克替尼125 mg,每日3次,持續1.5年治療。主要終點為無病生存期(DFS)。
2014年3月至2018年1月,共納入79例患者。中位隨訪時間為39.7個月,中位DFS和總生存期(OS)分別為41.4個月(95% CI: 33.6-51.8)和67.0個月(95% CI: 21.2-未達到[NR])。1年、3年、5年OS率分別為100%、83.3%、61.7%。BIM突變型和野生型患者的中位DFS無顯著差異(NE vs. 41.7個月;p = 0.75)。19 DEL和21 L858R的中位DFS無統計學意義。
埃克替尼作為輔助治療,在IIA-IIIA期EGFR突變非小細胞肺腺癌中顯示了良好的生存獲益,這與其他埃克替尼的研究一致。對于IIA-IIIA EGFR突變型非小細胞肺腺癌患者,R0切除后建議接受埃克替尼輔助治療,可進一步改善DFS,降低復發風險。此外,埃克替尼的最佳輔助時間仍不清楚,需要更多的數據來回答。
02
恩沙替尼MET 14跳躍突變探索
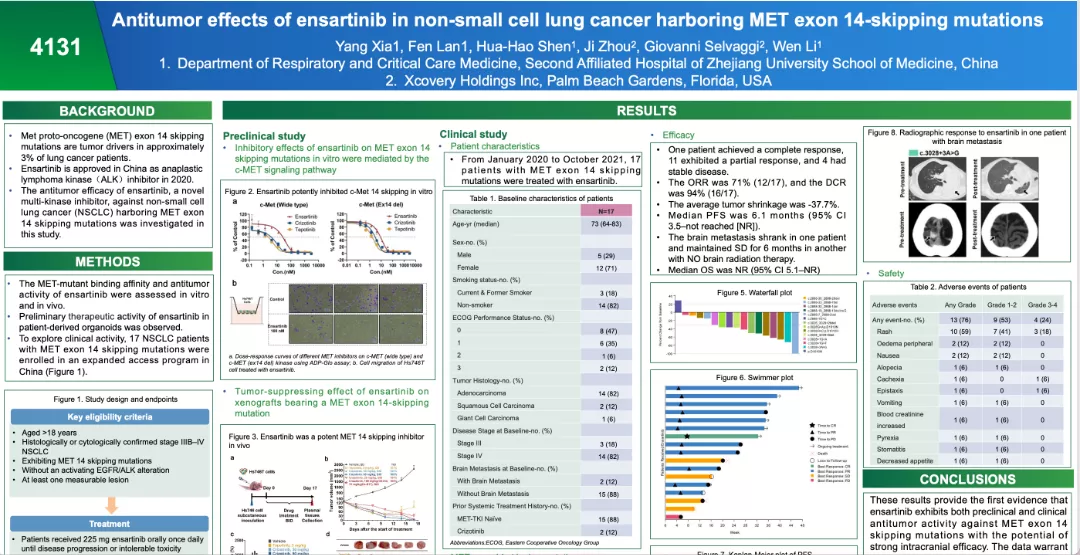
本次報道的數據主要由兩部分組成,一部分是臨床前研究數據,另一部分17名MET 14跳躍突變NSCLC的患者在一項同情使用方案下接受了恩沙替尼治療數據。
臨床前研究數據:Hs746T細胞增殖的影響試驗結果顯示恩沙替尼以濃度依賴性的方式抑制MET及其下游信號蛋白AKT和ERK的磷酸化,從而有效地抑制了具有MET第14外顯子跳躍突變的細胞增殖和遷移;Hs746T異種移植模型中恩沙替尼在不同劑量(30mg/kg、100mg/kg)治療組均顯示出明顯的腫瘤消退,同時與血漿相比,恩沙替尼在腫瘤、肝臟、肺和股骨中富集。另外,恩沙替尼腦內濃度(267 nM)遠高于體外激酶IC50值(7.9 nM),提示在動物模型和患者中,恩沙替尼可能抑制腦轉移。
臨床數據:從2020年1月1日至2021年10月31日,共有17名MET 14外顯子跳躍突變患者在一項同情使用方案下接受了恩沙替尼處方。17名患者中位年齡為73歲,其中15例患者為MET-TKI初治,2例為克唑替尼經治患者,17名MET14外顯子跳躍突變患者分別具有14種不同的突變類型。治療效果方面1例獲得完全緩解(CR),11例表現為部分緩解(PR), 4例病情穩定(SD)。17名患者ORR為71%(12/17), 疾病控制率(DCR)為94%(16/17),在15例TKI-naive的患者中ORR達80%(12/15)。中位PFS為6.1個月(95% CI 3.5–NR),中位OS仍未達到。安全性方面沒有因嚴重不良反應導致治療終止或死亡事件發生。
肺癌中MET通路失調通過多種機制發生,包括基因突變、擴增、重排和蛋白過表達。MET14外顯子跳躍突變在NSCLC中占比約為2-4%,是有前景的治療靶點。本次恩沙替尼MET 14外顯子跳躍突變探索初步成果證明恩沙替尼具有強效的抗腫瘤作用。目前恩沙替尼治療MET14跳躍突變非小細胞肺癌患者的II期臨床研究正在開展中,期待更多臨床證據的生成,造福MET 14外顯子跳躍突變的非小細胞肺癌患者。
03
BPI-361175,一種用于治療非小細胞肺癌的4代EGFR抑制劑

EGFR敏感突變(Del19和L858R)是非小細胞肺癌(NSCLC)的主要驅動突變(亞裔中占40%以上,非亞裔占10~20%)。EGFR抑制劑顯著延長攜帶EGFR突變NSCLC患者的PFS,提高他們的生活質量。然而,耐藥突變不可避免發生,導致疾病進展。EGFR C797S突變是3代EGFR抑制劑耐藥的主要原因,目前尚無治療策略。貝達自主研發的BPI-361175,是一種4代EGFR抑制劑,可用于治療EGFR的耐藥突變。
BPI-361175能夠顯著抑制多種不同突變EGFR激酶的活性以及攜帶相應突變的細胞EGFR磷酸化和細胞增殖,包括三突變(EGFRdel19 or L858R/T790M/C797S)、雙突變(EGFRdel19 or L858R/T790M、EGFRdel19 or L858R/C797S)和單突變(EGFRdel19 or L858R)。同時BPI-361175在細胞和激酶水平上均對EGFRWT和IGF1R無明顯抑制,體現出良好的選擇性。口服BPI-361175能夠抑制多種不同EGFR突變腫瘤的生長并使其消退,包括BaF3 EGFRDel19 or L858R/T790M/C797S、BaF3 EGFRDel19/C797S、PC9 EGFRDel19/T790M/C797S、NCI-H1975 EGFRDel19/T790M/C797S、NCI-H1975(EGFRL858R/T790M)、HCC827(EGFRDel19)等CDX模型和LDI-0025-200717(EGFRDel19/T790M/C797S)PDX模型。值得注意的是,BPI-361175具有良好的腦穿透能力,可以顯著延長BaF3 EGFRDel19/T790M/C797S腦原位模型小鼠的生存期。
總之,BPI-361175是一種強效、高選擇性、可廣泛覆蓋多種EGFR突變的口服4代EGFR抑制劑,能用于現有EGFR TKI治療NSCLC后的耐藥突變,并有望用于前線治療。BPI-361175在中國的臨床Ⅰ期試驗正在進行中,在美國的臨床Ⅰ期試驗預計在2022年第二季度開展。
04
BPI-371153,口服小分子PD-L1抑制劑
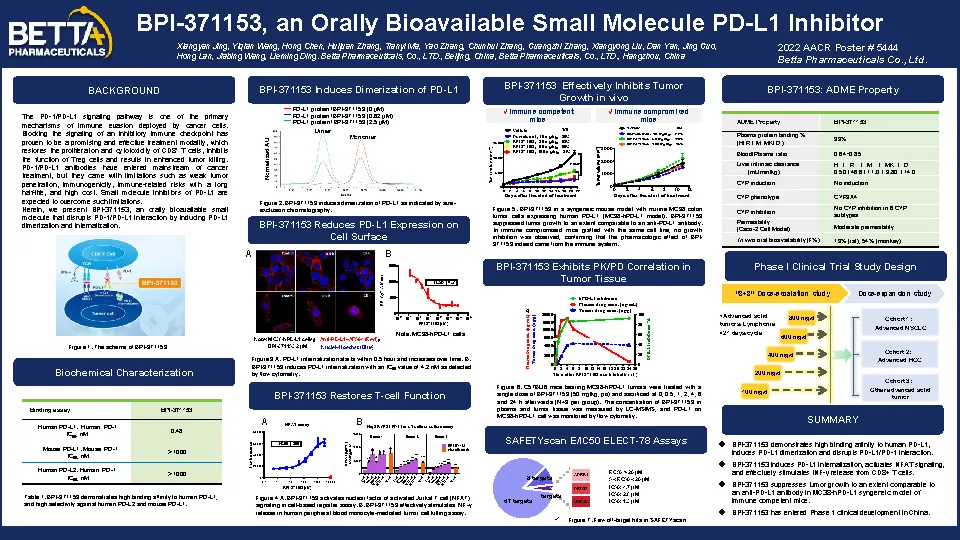
PD-1/PD-L1抗體已進入癌癥治療的主流,但也存在一些局限性,如腫瘤組織穿透性弱、免疫原性問題、客觀緩解率偏低、洗脫半衰期長等免疫相關的不良反應,以及成本高,依從性低等,而PD-L1的小分子抑制劑有望克服這些缺陷。BPI-371153是由貝達藥業股份有限公司自主研發的新分子實體化合物,屬于口服小分子PD-L1抑制劑,可有效誘導和穩定PD-L1二聚體的形成及內吞,從而強有力地阻斷PD-L1/PD-1蛋白-蛋白的相互作用。
體外實驗顯示,BPI-371153對人源PD-L1(hPD-L1)具有強效活性,可有效誘導hPD-L1二聚和內吞,阻斷PD-1/PD-L1的結合,從而解除PD-1的免疫抑制,激活T細胞下游信號通路,促進細胞因子IFN-γ的釋放。體內實驗顯示,在免疫系統功能完全的hPD-L1轉基因C57BL/6小鼠,荷瘤細胞穩定轉染過表達hPD-L1的小鼠結腸癌MC38細胞系模型上,BPI-371153抑制腫瘤生長的程度與抗PD-L1抗體近似;另外,在免疫系統功能缺陷的Balb/c 裸鼠模型上, BPI-371153未表現抑制腫瘤生長的作用,表明BPI-371153的抑瘤藥效是通過免疫系統實現的。此外,該化合物也具有良好的ADME性質,在臨床前多種動物種屬上具有較高的口服藥物暴露量。
BPI-371153作為一種新型口服小分子PD-L1抑制劑,已于2022年1月獲得中國NMPA的IND批準,目前正準備在局部晚期或轉移性實體瘤或復發/難治性淋巴瘤患者中開展Ⅰ期臨床試驗。
05
BPI-421286,靶向KRASG12C突變的強效小分子抑制劑
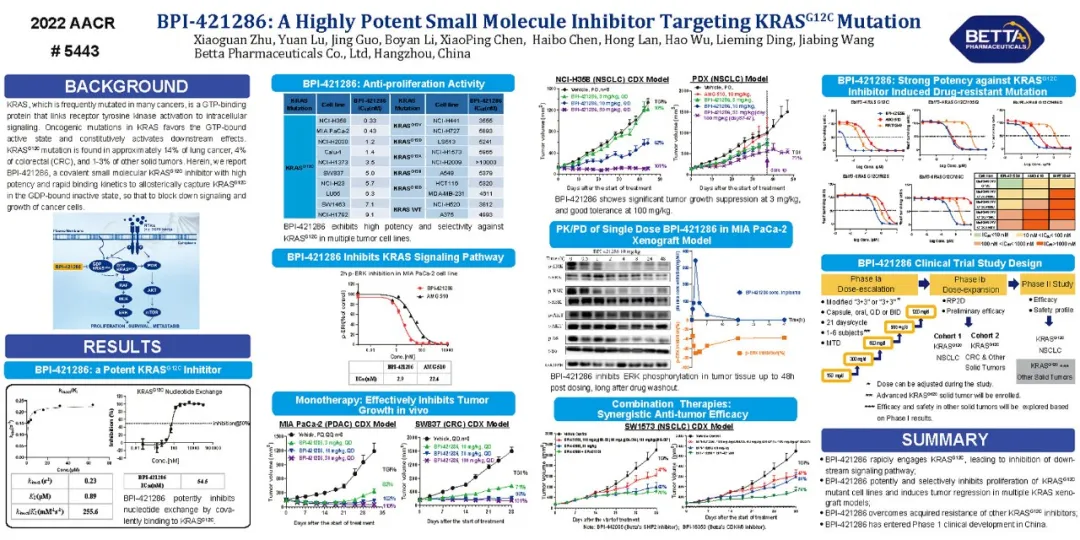
RAS是人類腫瘤中突變頻率最高的基因,在所有腫瘤中突變頻率占20-30%。KRASG12C是最常見的突變位點之一,在非小細胞肺癌中發生率約為14%,在結直腸癌發生率約為4%,在胰腺癌中發生率約為2%。KRAS過去三十多年一直被認為是一個“不能成藥”的蛋白靶標,缺乏有效治療手段,具有極大的未被滿足的醫療需求。近年來陸續有針對G12C位點突變的KRAS變構抑制劑進入臨床和獲批上市。貝達自主研發的BPI-421286是一種全新結構的強效,高選擇性且可能克服耐藥突變的共價小分子KRASG12C抑制劑。
在分子水平上,BPI-421286可迅速與KRASG12C蛋白形成共價鍵,具有較高的Kinact/KI值(256 mM-1 s-1)。細胞活性方面,BPI-421286對KRASG12C突變細胞系顯示出很強的增殖抑制作用,而對非KRASG12C突變以及野生型的細胞系無明顯抑制作用,體現出良好的選擇性;另外,BPI-421286能夠劑量依賴地抑制KRAS下游ERK蛋白的磷酸化。在體內,每日口服BPI-421286能夠抑制多種KRASG12C突變的異種移植瘤(CDX)或PDX模型的腫瘤生長,甚至使其消退,包括非小細胞肺癌、結直腸癌和胰腺癌。另外,BPI-421286與多種靶向藥物(如SHP2抑制劑BPI-442096和CDK4/6抑制劑BPI-16350)聯合使用后,均顯示出比單藥更強的抑制腫瘤生長的作用。更重要的是,對于KRASG12C抑制劑治療后臨床觀察到的獲得性耐藥突變,如G12C/H95D、G12C/H95Q、G12C/R68S、G12C/Y96C,BPI-421286也表現出較強的抑制活性,表明BPI-421286有望克服已上市的KRASG12C抑制劑引發的獲得性耐藥。
總之,BPI-421286是一種高活性、高選擇性的共價小分子KRASG12C抑制劑,具有良好的ADME特性,臨床前毒理學研究中顯示出較大的安全窗。BPI-421286正在中國開展Ⅰ期臨床研究。
06
BPI-442096,可治療多種腫瘤的強效高選擇性的SHP2抑制劑
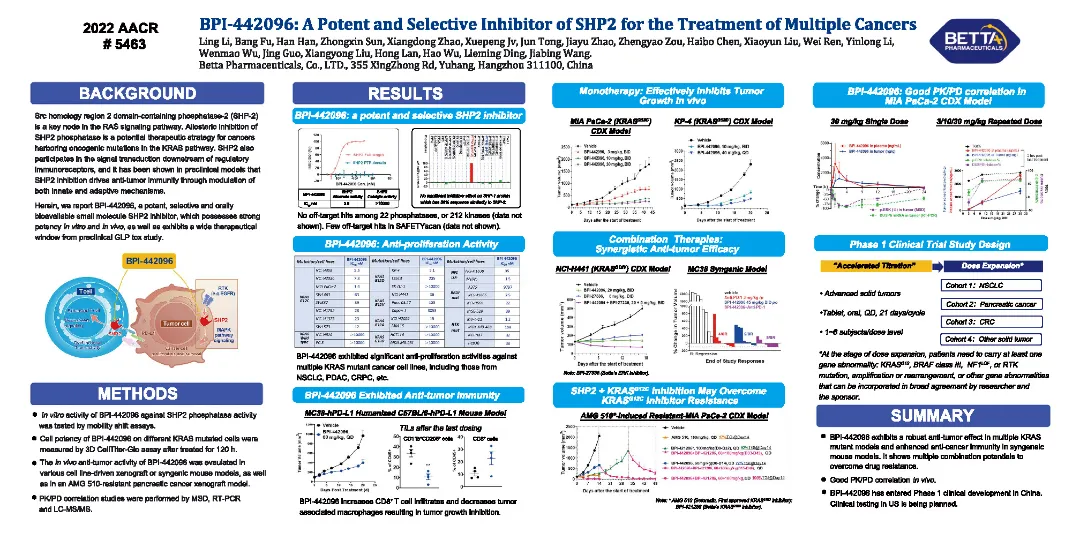
含Src 同源2結構域蛋白酪氨酸磷酸酶(SHP2)作為腫瘤RAS信號通路的關鍵調控節點,變構抑制其活性是治療多種腫瘤的潛在治療策略;同時,SHP2參與腫瘤免疫抑制性信號傳導。臨床前研究表明,靶向SHP2可發揮直接抑制腫瘤細胞增殖以及恢復或增強抗腫瘤免疫應答的雙重功能。
貝達自主研發的BPI-442096是一種強效、高選擇性、可口服的SHP2抑制劑。BPI-442096對多種KRAS突變腫瘤細胞,包括非小細胞肺癌、胰腺導管癌、結直腸癌等均表現出顯著增殖抑制活性。BPI-442096可濃度依賴性地抑制SHP2磷酸酶活性、腫瘤細胞下游ERK磷酸化水平、以及免疫細胞PD-1/PD-L1信號下游NFAT報告基因表達。在多種攜帶KRASG12C、KRASG12D、KRASG12V突變的小鼠異種移植瘤模型,BPI-442096均表現出顯著的體內腫瘤生長抑制作用;在MC38小鼠同種移植瘤模型中,BPI-442096單藥或聯用PD-1/PD-L1靶向藥物同樣發揮抗腫瘤生長的作用。此外,研究表明BPI-442096可發揮協同增強KRASG12C抑制劑對獲得性耐藥腫瘤的作用,克服KRASG12C抑制劑獲得性耐藥。臨床前研究結果顯示,BPI-442096在多種屬上有較高的口服生物利用度,及合適的ADME性質和安全性。
總之,BPI-442096在多種KRAS突變腫瘤模型、同種腫瘤模型上發揮顯著抗腫瘤藥效,同時展現出聯合用藥克服耐藥的潛力。BPI-442096正在中國進行臨床Ⅰ期研究。
下一條: 貝達藥業鹽酸恩沙替尼膠囊術后輔助治療獲批開展臨床試驗
熱門點擊